
Imara’s IMR-687 for SCD Receives FDA Rare Pediatric Disease Designation



The U.S. Food and Drug Administration (FDA) granted rare pediatric disease designation to IMR-687, Imara’s drug candidate for the treatment of sickle cell disease (SCD). The investigational therapy has also been granted orphan drug status by the FDA.
Rare pediatric disease designation is granted to drugs that show promise for rare and serious or life-threatening diseases in U.S. patients, primarily age 18 years or younger. The designation provides incentives to advance the development of rare disease drugs, including access to the FDA’s expedited review and approval programs.
“People living with sickle cell disease have limited treatment options for its devastating effects,” James McArthur, PhD, Imara’s president and CEO, said in a news release. “The FDA’s decision reflects its commitment to working with innovators and the patient community toward advancing safe and effective treatments for children suffering from the rare and damaging pediatric manifestations of the disease.”
McArthur said the biotech company is looking forward to “working closely with the FDA throughout our IMR-687 clinical program, as this is important validation of Imara’s work in developing promising therapeutics to address serious medical needs for patients living with sickle cell disease.”
IMR-687 is a highly potent, selective inhibitor of phosphodiesterase-9 (PDE9) in blood cells that reportedly reduces both red blood cell sickling and blockage of blood vessels, the underlying causes of SCD.
Previous studies on animal models have shown that IMR-687 increases fetal globin, the type of hemoglobin that is found in the human fetus and has the ability to bind oxygen with higher affinity than adult hemoglobin. This increase reduces red blood cell sickling and, consequently, the death of red blood cells, thereby preventing blood vessel blockade.
A Phase 1a clinical study (NCT02998450) is ongoing and recruiting healthy volunteers to assess the safety of IMR-687 as well as the drug’s behavior in the body. Depending on the results obtained, Imara will start a Phase 2a study with adult sickle cell disease patients later in 2017, and a Phase 2 study with pediatric patients in 2018.
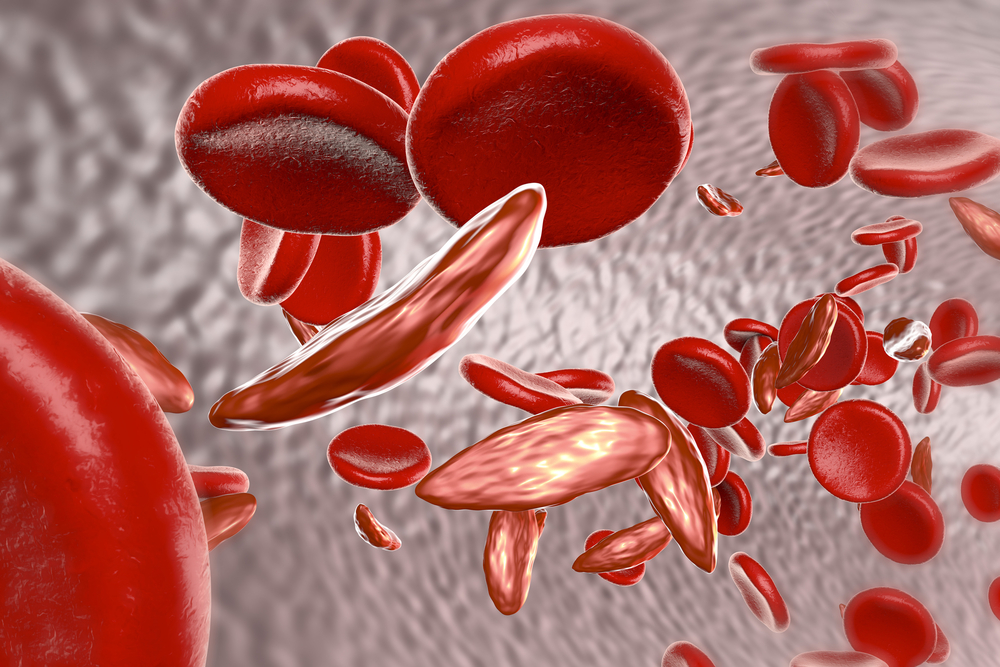
SCD is a genetic disorder characterized by morphological changes occurring in the oxygen transporting protein hemoglobin, which travels in red blood cells. The affected red blood cells have a distorted sickle-like shape, which makes these cells stick together, raising the risk of blood vessel blockade.
The faulty red blood cells also have a reduced ability to carry oxygen compared to normal red blood cells. This can lead to lack of oxygen delivery to tissues and cause permanent damage to organs, including the liver, spleen, kidneys and brain.


