Findings on Abnormal Hemoglobin Assembly Could Lead to Lower-Dose SCD Medications, Researchers Say
shutterstock



A study of sickle cell disease (SCD) at the molecular level found abnormal hemoglobin molecules assemble much faster and less efficiently than previously thought — findings that could lead to the development of new treatments.
The researchers said better understanding of the assembly process is the first step toward developing new medicines that would be effective at lower doses and would cause fewer side effects for patients.
The study, “Rapid and inefficient kinetics of sickle hemoglobin fiber growth,” was published in the journal Science Advances.
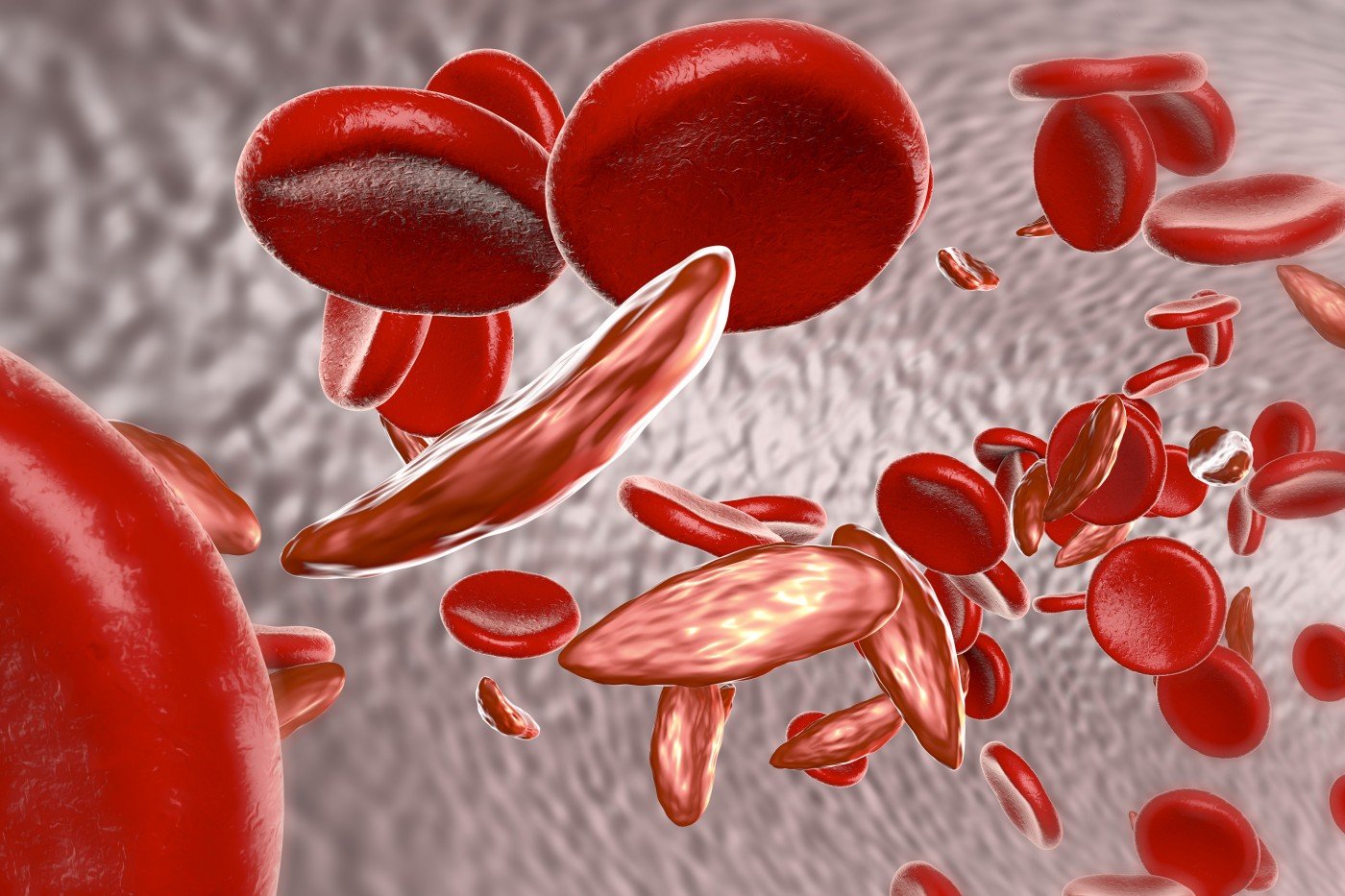
SCD is a rare genetic disorder caused by mutations in the HBB gene, which provides instructions for making hemoglobin, a protein responsible for transporting oxygen in the blood. These mutations change the structure of the protein, resulting in the production of abnormal hemoglobin fibers. These fibers then tend to stiffen red blood cells, changing their shape from a normal disc into a sickle-like shape.
These abnormal red blood cells, which are less efficient at transporting oxygen through the body, also tend to stick to each other and form blood clots that block blood circulation. This prevents the delivery of normal nutrients and oxygen to tissues, and causes one of the disease’s most common and painful complications, vaso-occlusive crisis (VOC). It also increases the risk of stroke and infection.
“Even though it has been known for decades what causes sickle cell disease at the molecular level, no one has ever studied the disease at this level of detail,” David Wood, PhD, an associate professor of biomedical engineering at the University of Minnesota and a senior author of the study, said in a news release.
“What we found at the nanoscale was quite surprising. We found that the disease self-assembly process is less efficient than we thought, which means that it could be easier to develop new medicines that would be effective at lower doses and would cause fewer side effects for patients,” he said.
Researchers from the University of Minnesota College of Science and Engineering analyzed the assembly mechanism of abnormal hemoglobin fibers using high-resolution microscopes and cameras that allowed the visualization of these structures at the nanoscale level.
According to their findings, the self-assembly process of sickle hemoglobin is much faster and less efficient than previously thought, with only 4% efficiency instead of the 96% that other, earlier studies found.
“It’s kind of like building a tower by stacking LEGOs,” Wood said. “For every 100 LEGOs only four actually stay on as part of the tower. I would have to stack another 100 LEGOs to get another four to stay on. This shows us that this is a much more inefficient process than previously thought and that it wouldn’t take as much medicine to disrupt this process.”
Based on their calculations, the researchers believe a compound would only need to bind to approximately 5% of the total abnormal hemoglobin molecules in circulation to be effective in blocking the assembly of sickle hemoglobin fibers.
“It may be possible to treat affected individuals with compounds at concentrations that are only a fraction of the total amount of hemoglobin,” the researchers said.
With only two medications — Endari (L-glutamine) and hydroxyurea — currently approved by the U.S. Food and Drug Administration to treat SCD patients, Wood and his colleagues are confident these findings may open the door to new treatment options capable of helping the millions of people worldwide affected by the disease.
“Our results indicate that targeting (…) the fiber growth process with drug treatment has the potential to be a viable approach for more effective treatment of sickle cell disease, and we are optimistic that this demonstration of feasibility may spur development of much needed antisickling therapies,” they said.