
White Blood Cells Regulate Levels of Fetal Hemoglobin in Sickle Cell Patients, Study Shows
Written by |
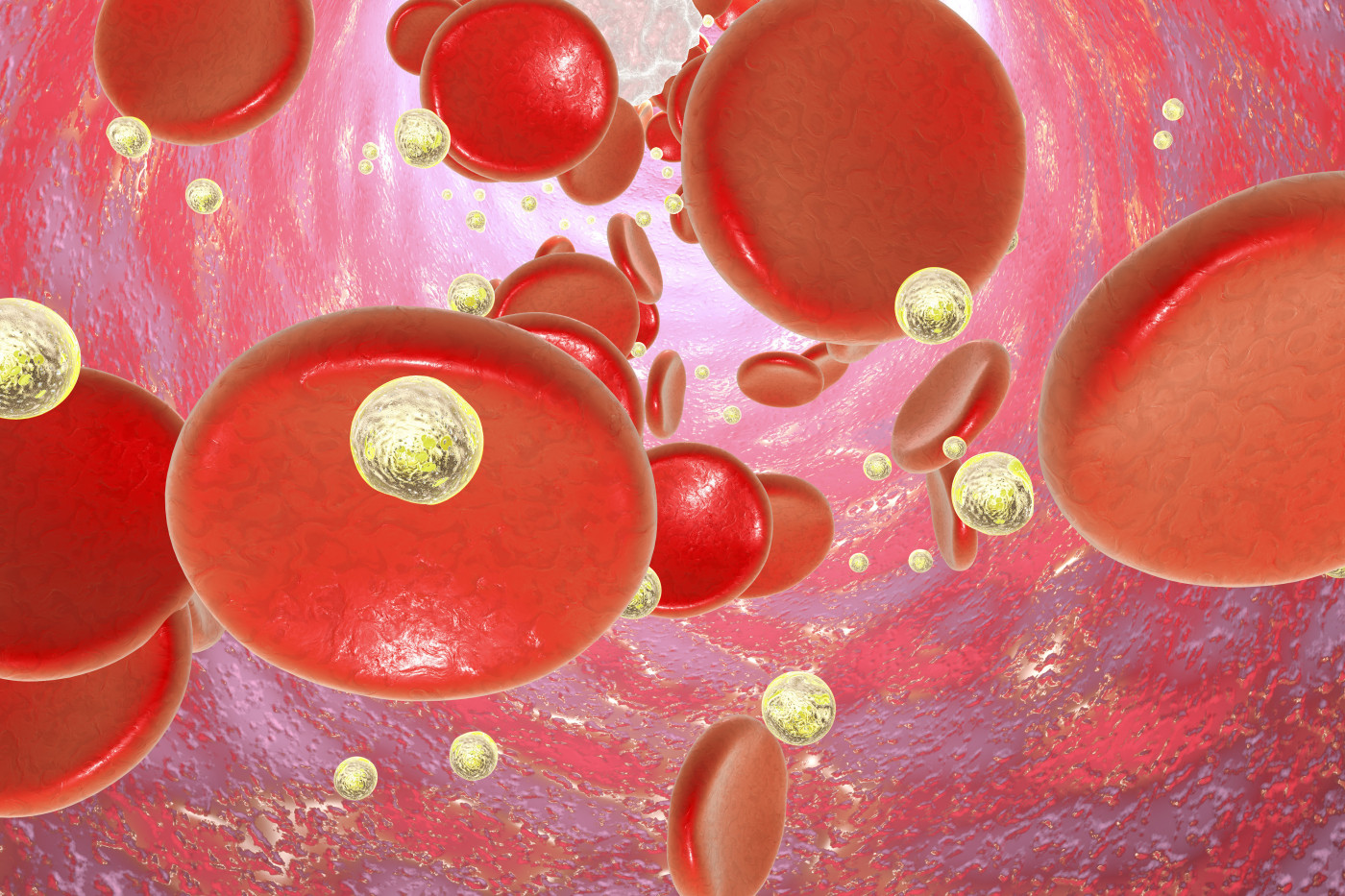
White blood cells were seen to produce molecules that decrease a specific type of hemoglobin — the protein in red blood cells that’s responsible for the transport of oxygen — in patients with sickle cell disease (SCD).
These findings may help in the understanding of disease mechanisms, as well as resistance to treatment.
Researchers confirmed the inverse correlation between the number of white blood cells and the production of this type of hemoglobin, called fetal hemoglobin, and the negative impact of these molecules on fetal hemoglobin production.
Researchers from the Medical College of Georgia, Augusta University, in Augusta, Georgia, published the data in a study titled “Serum of sickle cell disease patients contains fetal hemoglobin silencing factors secreted from leukocytes,” in the Journal of Blood Medicine.
Fetal hemoglobin is responsible for carrying the oxygen in the human fetus from the second month of development in the uterus until the newborn reaches six months old.
An increase in fetal hemoglobin production caused by medication, specifically hydroxyurea, is known to prevent SCD symptoms.
Previous studies have demonstrated that sickle cell patients with high levels of fetal hemoglobin have a reduced number of white blood cells, suggesting that these immune cells play a role in the regulation of fetal hemoglobin in the disease.
Elevated numbers of white blood cells have also been associated with risk of early death, as well as clinical complications later in life.
To further study a potential correlation between these factors, the research team investigated the presence of molecules capable of silencing the production of fetal hemoglobin in the blood of sickle cell patients receiving therapy.
The team retrospectively collected data on 337 adult SCD patients, 82 of whom were being treated with hydroxyurea, a medicine that stimulates the production of fetal hemoglobin, in a daily dose of 15 to 35 mg per kg body weight of patients.
Hydroxyurea treatment was seen to increase not only the production of fetal hemoglobin, but also the levels of total hemoglobin and to improve red blood cell parameters.
The data collected from patients confirmed that the number of white blood cells, specifically neutrophils and lymphocytes/monocytes, decreased when the levels of fetal hemoglobin increased, indicating that these cells may play a role in hydroxyurea-mediated fetal hemoglobin production.
Lab studies in stem cells showed that the patients’ blood contains molecules capable of hampering the production of fetal hemoglobin.
Researchers found that the effect of these molecules can be reduced with hydroxyurea treatment.
With this study, researchers were able to demonstrate the importance of white blood cells in the regulation of fetal hemoglobin levels for sickle cell disease patients, regardless of their medication status.
“This study has shown that hydroxyurea-induced fetal hemoglobin expression is regulated at least in part by the mechanisms controlling leukocyte counts, and that fetal hemoglobin silencing factors that are secreted, possibly by leukocytes, may be involved in hydroxyurea-regulated fetal hemoglobin expression in SCD,” researchers wrote.
“Further insight into [fetal hemoglobin] silencing factors might help us clarify the mechanisms underlying hydroxyurea resistance as
seen in a subset of SCD patients,” they added.

Leave a comment
Fill in the required fields to post. Your email address will not be published.