
Link Found in Iron Overload with Sickle Cell Disease, Genetic Mutations
Written by |
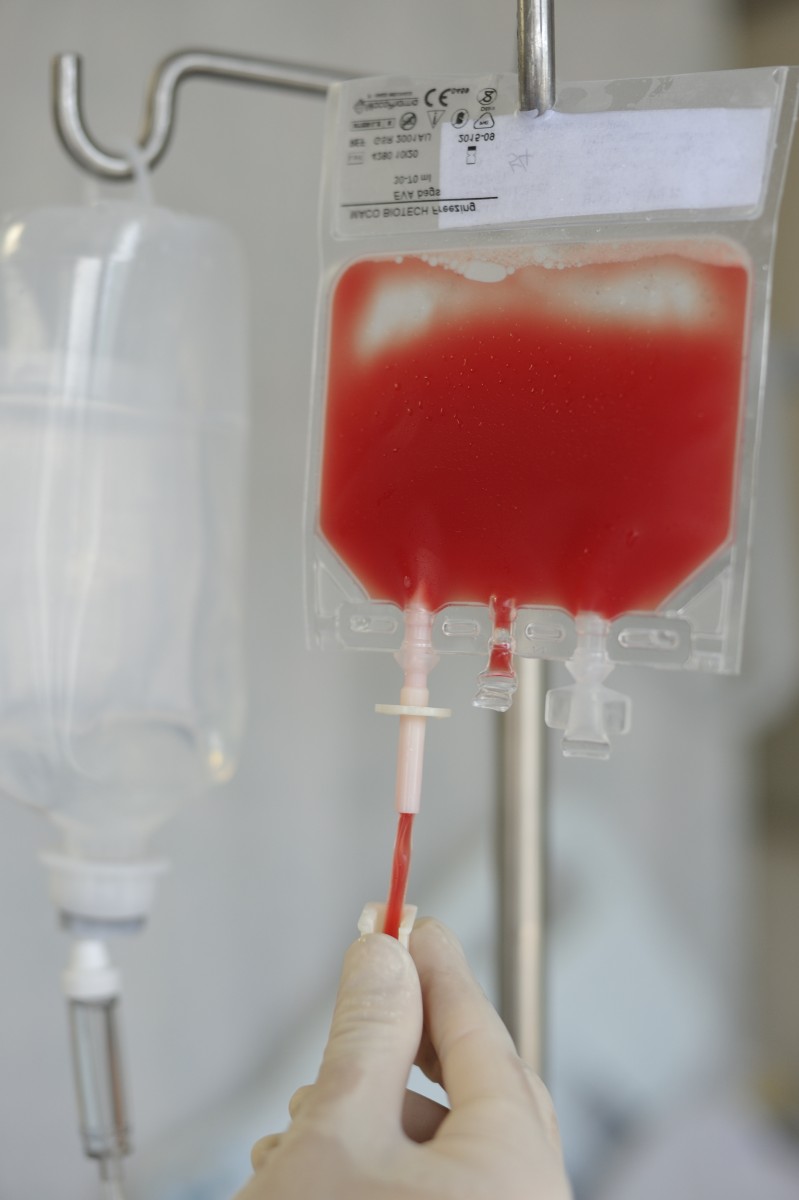
A study performed by researchers at Baskent University, in Ankara, Turkey, found that mutations in certain genes play a role in the deposition of iron in the liver of patients with sickle cell (SC) disease.
Findings, presented in the study “Effect of Hereditary Hemochromatosis Gene (HFE) H63D and C282Y Mutations on Iron Overload in Sickle Cell Disease Patients,” were published in the Turkish Journal of Hematology.
SC, also called sickle cell anemia, is a hereditary disease characterized by rigid and sickle-like shaped hemoglobin, a protein molecule found in red blood cells responsible for carrying oxygen from the lungs to the body’s cells which return formed carbon dioxide from cells back to the lungs. The sickle-shaped cellular deformation reduces the oxygen supply throughout the body’s cells and results in several health problems including haemolytic crises, where the hemoglobin level drops at accelerated rates. The problem is usually managed by blood transfusions, but a repeated supply of red blood cells may lead to hemochromatosis, a condition of iron overload that results in mortality and morbidity of SC anemia patients.
SC is caused by a mutation in a gene called human hemochromatosis protein (HFE). Data concerning the role of gene mutation in iron overload and its deposition (storage) in tissues are lacking. In the Baskent University study, researchers examined the influence of mutations in the HFE gene, specifically p.H63D and p.C282Y, on transfusion-related cardiac and liver iron overload in 44 SC patients between 2008 and 2013.
The patients were divided into two main groups. The first group (group A) was administered a chelation therapy to allow the capture of free iron in blood and prevent its storage in the liver. The second group (group B) received no treatment. Iron levels in the blood of the patients were then analyzed directly and indirectly through magnetic resonance imaging (MRI) and biochemistry. Later, the HFE gene mutations were examined by standard methods and statistical analyses were performed.
Examination of the results revealed the presence of p.H63D mutation in 32.3% of patients of the first group and 7.7% patients in the second group. Comparison of iron overload between the groups showed significant iron storage in the liver of patients of group B. In group A, patients with HFE mutation had a higher iron storage when compared to those without the mutation.
Authors of the study concluded that HFE gene mutations are important in iron deposition in the liver in patients with sickle cell disease; a determination that could help to improve treatment of the disease over the long-term.


Leave a comment
Fill in the required fields to post. Your email address will not be published.